Taking the Guesswork out of HFpEF
With an aging population and a higher burden of comorbidities, the proportion of heart failure patients with a preserved ejection fraction, i.e. ejection fraction ≥ 50% is increasing.1 Heart failure with preserved ejection fraction (HFpEF) now accounts for more than half of all heart failure hospitalizations. Despite the increasing prevalence, HFpEF remains a nebulous entity. HFpEF is often alluded to without a complete understanding of the underlying pathophysiology. Diagnosing HFpEF can be challenging as opposed to heart failure with reduced ejection fraction (HFrEF). With a normal ejection fraction, attributing dyspnea to cardiac congestion without performing invasive hemodynamic testing requires good clinical suspicion and judgment. Moreover, euvolemic patients with compensated HFpEF can have elevated filling pressures and dyspnea only with exertion. Non-invasive measurement of cardiac pressures can be inconclusive and invasive cardiopulmonary exercise testing (CPET) is considered the gold standard in this population.2 However its routine use is not feasible as it is an invasive and technically complex procedure with limited availability. An algorithm incorporating clinical and non-invasive parameters can help stratify patients’ probability of having HFpEF. The utility of risk scores, such as the CHA2DS2-VASc, TIMI, and Wells’ scores, is well established in the field of cardiovascular disease. There has been research into developing similar algorithms/ prediction scores for the diagnosis of HFpEF. Here, we discuss 2 proposed scoring systems for HFpEF- H2FPEF and HFA-PEFF.
H2FPEF Score
Reddy and colleagues at Mayo Clinic, Rochester developed the H2FPEF score to help physicians discriminate HFpEF from non-cardiac causes of dyspnea in symptomatic patients without obvious fluid overload.3 The score was developed from a cohort of 414 patients with an ejection fraction ≥ 50%, who underwent invasive hemodynamic exercise testing for definitive evaluation of unexplained dyspnea. Different clinical and echocardiographic markers were evaluated through logistic regression to identify variables associated with HFpEF. Ultimately 6 routinely available variables (BMI > 30 kg/m2, atrial fibrillation, hypertension treated with ≥ 2 medications, pulmonary artery systolic pressure > 35 mmHg, age > 60 years, and E/e’ > 9) were used for the model. Each variable was assigned a point based on the strength of association observed with HFpEF diagnosed via invasive testing (Figure 1). The final score had good discriminatory power (area under the curve = 0.84) for differentiating HFpEF from other causes of dyspnea. As the score increased from 0 to 9, so did the probability of HFpEF. The robustness of the model was validated through sensitivity analyses and a test cohort of 100 patients. The authors proposed a Bayesian approach- using a low score (0-1) to rule out HFpEF, a high score (6-9) to make a diagnosis of HFpEF, and an intermediate score (2-5) to consider additional testing.
The major limitation of this important model is the setting of the study. It was conducted at a single institute serving as a referral center, which may not truly represent the general population. It is reassuring that the score has been validated in small external cohorts.4,5 Moreover, an analysis from the TOPCAT trial population showed that patients with a higher H2FPEF score had an increased risk of adverse outcomes, suggesting a prognostic value of the score.6
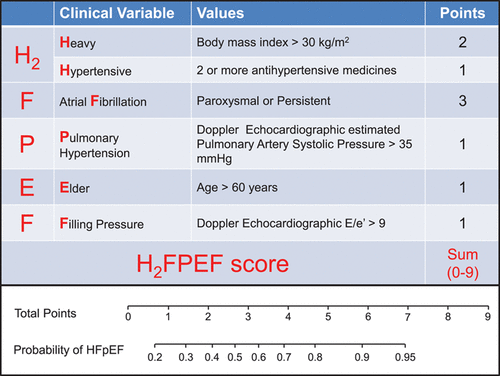
Figure 1. H2FPEF Score proposed by Reddy et al. https://doi.org/10.1161/CIRCULATIONAHA.118.034646
HFA-PEFF Score
In 2020, the Heart Failure Association (HFA) and the European Society of Cardiology (ESC) released a consensus recommendation for diagnosing HFpEF, proposing the stepwise HFA-PEFF algorithm.7 This recommendation centers around the use of the HFA-PEFF score. The proposed scoring system uses echocardiographic parameters and natriuretic peptide (BNP and NT-proBNP) levels. The variables are divided into major and minor criteria across 3 domains- functional, morphological, and biomarker (Figure 2). Parameters within a domain are not additive, hence the score can be used even when certain values are not available. Each domain can contribute a maximum of 2 points and the total score ranges from 0 to 6. A total score of 5-6 is considered diagnostic of HFpEF while a score of 0-1 makes HFpEF unlikely. An intermediate score of 2-4 warrants further testing with non-invasive or invasive functional testing.
Notably, the HFA-PEFF score did not utilize demographic and clinical parameters and the power of the score was not assessed. However, an independent study later demonstrated its validity in two separate cohorts.8
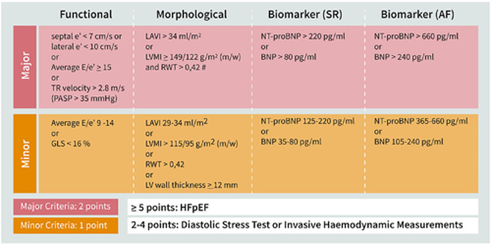
Figure 2. HFA-PEFF Score proposed by HFA and ESC. https://doi.org/10.1002/ejhf.1741
Both of the above scoring systems have been received with enthusiasm, given the lack of a clear definition and diagnostic framework for HFpEF. Studies evaluating the 2 scores have also been published. Parcha and colleagues studied the generalizability of the H2FPEF and HFA-PEFF scores in an analysis of participants with unexplained dyspnea from prior HFpEF trials and the Atherosclerosis Risk in Communities (ARIC) study.9 They found that both the scores could rule out HFpEF with a greater than 99% success rate but the H2FPEF score had a higher specificity than the HFA-PEFF score. Amanai and colleagues calculated H2FPEF and HFA-PEFF scores in patients with HFpEF referred for stress echocardiography.10 They found that both scores had similarly high positive and negative predictive values and a correlation with abnormal hemodynamics during exercise. Another study in the ARIC population also found that both high H2PEFF and HFA-PEFF scores were associated with increased risk of heart failure hospitalizations or death, suggesting the prognostic value of both.11
Both H2FPEF and HFA-PEFF are validated and easy-to-use scores using readily available clinical, laboratory, and echocardiographic parameters. The use of these scores in the appropriate patient and context can aid in the timely and accurate diagnosis of HFpEF. Growing recognition and emergence of effective therapies such as SGLT2 inhibitors are important strides for improving outcomes for patients with HFpEF.
References:
- Borlaug BA. Evaluation and management of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2020;17(9):559-573. doi:10.1038/s41569-020-0363-2
- Sorajja P, Borlaug BA, Dimas VV, et al. SCAI/HFSA clinical expert consensus document on the use of invasive hemodynamics for the diagnosis and management of cardiovascular disease. Catheter Cardiovasc Interv. 2017;89(7):E233-E247. doi:10.1002/ccd.26888
- Reddy YNV, Carter RE, Obokata M, Redfield MM, Borlaug BA. A Simple, Evidence-Based Approach to Help Guide Diagnosis of Heart Failure With Preserved Ejection Fraction. Circulation. 2018;138(9):861-870. doi:10.1161/CIRCULATIONAHA.118.034646
- Sepehrvand N, Alemayehu W, Dyck GJB, et al. External Validation of the H2F-PEF Model in Diagnosing Patients With Heart Failure and Preserved Ejection Fraction. Circulation. 2019;139(20):2377-2379. doi:10.1161/CIRCULATIONAHA.118.038594
- Segar MW, Patel KV, Berry JD, Grodin JL, Pandey A. Generalizability and Implications of the H2FPEF Score in a Cohort of Patients With Heart Failure With Preserved Ejection Fraction. Circulation. 2019;139(15):1851-1853. doi:10.1161/CIRCULATIONAHA.118.039051
- Myhre PL, Vaduganathan M, Claggett BL, et al. Application of the H2 FPEF score to a global clinical trial of patients with heart failure with preserved ejection fraction: the TOPCAT trial. Eur J Heart Fail. 2019;21(10):1288-1291. doi:10.1002/ejhf.1542
- Pieske B, Tschöpe C, de Boer RA, et al. How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail. 2020;22(3):391-412. doi:10.1002/ejhf.1741
- Barandiarán Aizpurua A, Sanders-van Wijk S, Brunner-La Rocca HP, et al. Validation of the HFA-PEFF score for the diagnosis of heart failure with preserved ejection fraction. Eur J Heart Fail. 2020;22(3):413-421. doi:10.1002/ejhf.1614
- Parcha V, Malla G, Kalra R, et al. Diagnostic and prognostic implications of heart failure with preserved ejection fraction scoring systems. ESC Heart Fail. 2021;8(3):2089-2102. doi:10.1002/ehf2.13288
- Amanai S, Harada T, Kagami K, et al. The H2FPEF and HFA-PEFF algorithms for predicting exercise intolerance and abnormal hemodynamics in heart failure with preserved ejection fraction. Sci Rep. 2022;12(1):13. doi:10.1038/s41598-021-03974-6
- Selvaraj S, Myhre PL, Vaduganathan M, et al. Application of Diagnostic Algorithms for Heart Failure With Preserved Ejection Fraction to the Community. JACC Heart Fail. 2020;8(8):640-653. doi:10.1016/j.jchf.2020.03.013
“The views, opinions, and positions expressed within this blog are those of the author(s) alone and do not represent those of the American Heart Association. The accuracy, completeness, and validity of any statements made within this article are not guaranteed. We accept no liability for any errors, omissions, or representations. The copyright of this content belongs to the author and any liability with regards to infringement of intellectual property rights remains with them. The Early Career Voice blog is not intended to provide medical advice or treatment. Only your healthcare provider can provide that. The American Heart Association recommends that you consult your healthcare provider regarding your health matters. If you think you are having a heart attack, stroke, or another emergency, please call 911 immediately.”