Clinical Features and Pathophysiology of Transthyretin Cardiac Amyloidosis (TTR-CA)
There has been an increasing awareness of the presence of cardiac amyloidosis (CA) in patients presenting with heart failure with preserved left ventricular ejection fraction (HFpEF). The clinical features suggestive of CA are recurrent heart failure with preserved ejection fraction (HFpEF) and restrictive left ventricular filling of unclear etiology. These patients characteristically have concentric left ventricular hypertrophy with wall thickness greater than 12 mm, biatrial enlargement, increased thickness of the interatrial septum and left atrial dysfunction even in the absence of atrial fibrillation or atrial flutter1. Typically, these patients have abnormal global longitudinal strain with “apical sparing” pattern2.
Amyloidoses are a group of protein-folding disorders in which more than one organ is infiltrated by proteinaceous deposits known as amyloid. The deposits are derived from one of several amyloidogenic precursor proteins, and the prognosis of the disease is determined both by the organ(s) involved and the type of amyloid. Amyloid involvement of the heart, cardiac amyloidosis (CA), carries the worst prognosis of any involved organ, and light-chain amyloidosis (AL-CA) is the most serious form of the disease (1). CA may be due to myocardial deposition of transthyretin protein derived from the liver known as transthyretin cardiac amyloidosis (TTR-CA) or may be due to AL-CA with myocardial deposition of immunoglobulin light chain proteins derived from a clone of plasma cells1. This blog will focus on the treatment of TTR-CA after a brief discussion about diagnosing this disease.
Diagnosing Transthyretin Cardiac Amyloidosis (TTR-CA)
TTR-CA is an underrecognized etiology for patients with recurrent exacerbations of HFpEF. However, the use of bone avid radiotracers such as 99m- technetium pyrophosphate (99m-Tc PYP) to diagnose TTR-CA have changed the diagnostic paradigm of this disease and have improved the ability to diagnose the disease readily1. Typically once the diagnosis is suspected clinically and by echocardiography or MRI, these patients should undergo clonal analysis with serum and urine free light chains and serum and urine immunofixation1. In patients who undergo 99mTc-PYP scans and have grade 2 or 3 myocardial uptake of the radiotracer on SPECT imaging, the positive predictive value of this finding with negative clonal analysis is close to 100% often deferring the need for myocardial biopsy for these patients3. It is important that once the diagnosis of TTR-CA is made that these patients undergo genotyping to determine if the mutant form (TTR-CAm) is present which is hereditary and presents earlier, usually 40+ years of age and has a slight male predominance compared with female1. In patients with TTR-CA who are genotype negative, these patients are defined as having the “wild type” sporadic form of TTR-CA (TTR-CAwt) and are usually older at 65+ years of age with a significant male predominance of 15:11.
Emerging Therapeutic Agents for Transthyretin Cardiac Amyloidosis
With the increasing awareness of TTR-CA and the ability to diagnose this disease that was once difficult to diagnose, there have been the development of various treatment options for these patients. There are three potential targets for treatment of patients with TTR-CA. These three potential targets include suppression of TTR synthesis, TTR stabilization and TTR fibril degradation and absorption4 Figure 1.
(i) Suppression of Transthyretin (TTR) synthesis
TTR synthesis can be suppressed by liver transplantation4. However there are 2 potential therapeutic medications that have been studied and have been shown to decrease TTR-Synthesis4. These 2 agents that have been shown to decrease TTR synthesis are patisiran5 and inotersen6.
Patisiran was shown in the APOLLO clinical trial to improve multiple clinical manifestations of hereditary transthyretin amyloidosis (TTR-CAm)5.
Patisiran is a RNA interference therapeutic agent that specifically inhibits hepatic synthesis of transthyretin. In the APOLLO clinical trial patients with TTR-CAm with polyneuropathy were studied and were treated with intravenous aptisiran 0.3 mg per kilogram of body weight once every 3 weeks after randomization and were compared with patients treated with placebo. Patisiran was shown at 18 months to result in a sustained and rapid decrease in transthyretin serum levels in patients treated with this agent with a 81% mean reduction in serum levels for all ages, gender and genotypes. Patisiran halted or reversed the progression of transthyretin amyloidosis and reduced the related neuropathic symptoms. This agent also improved the ability to ambulate with regards to improved gait speed and mobility and there was also an improvement in quality of life. The cardiac manifestation of the disease with regards to the echocardiographic measures of cardiac structure and function improved and there was also a reduction in NT-proBNP levels. The safety profile of the medication was good with the only side effects of the drugs being described as an increased incidence of peripheral edema and mild to moderate infusion related reactions with patisiran use5. There were no hematologic or nephrotoxic side effects of Patisiran noted during the study5.
Inotersen was shown in the international, randomized, double-blind, placebo-controlled NEURO-TTR study to improve the course of neurologic disease and quality of life in patients with TTR-CA6. Inotersen is a 2′-O-methoxyethyl–modified antisense oligonucleotide that inhibits hepatic production of transthyretin. In the NEURO-TTR study adults with stage 1 (patient is ambulatory) or stage 2 (patient is ambulatory with assistance) with TTR-CAm with polyneuropathy were included in the study and were randomized in 2:1 fashion to receive weekly subcutaneous injections of inotersen (300 mg) or placebo. The study period was 15 months. Over this study period the course of the neurologic symptoms related to TTR-CAm improved in addition to the quality of life. Steady state levels in reduction of circulating transthyretin protein was reached within 13 weeks and was sustained throughout the study period. The mean nadir in the decrease in circulating transthyretin from baseline levels in the inotersen group was a mean nadir of 74%6. The significant side effects of inotersen are thrombocytopenia and glomerulonephritis, therefore this should be managed with enhanced monitoring and treatment6.
Both patisiran and inotersen were approved by the Food and Drug Administration (FDA) for treatment of patients with TTR-CAm with evidence of neuropathy.
(ii) TTR Stabilization
Tafamidis is a benzoxazole derivative lacking nonsteroidal antiinflammatory drug activity that binds to the thyroxine-binding sites of transthyretin with high affinity and selectivity and inhibits the dissociation of tetramers into monomers thus stabilizing the transthyretin protein in TTR-CA. Tafamidis was shown in the multicenter, international, double-blind, placebo-controlled ATTR-ACT study to be a TTR stabilizer that decreased all-cause mortality, cardiovascular related hospitalization rate and the decline in functional capacity in patients with TTR-CA7. In this study, patients were randomized in 2:1:2 fashion to 80mg of tafamidis or 20 mg of tafamidis or placebo over a 30 month period. Patients who received tafamidis had improved survival, had decreased cardiovascular (CV) related hospitalization rate and had less decline in functional capacity. This effect on decreased CV related hospitalization rate was seen amongst all patient treatment groups receiving tafamidis with the exception of those patients with New York Heart Association Class III heart failure. This was thought to be attributable to longer survival during the more severe stage of this disease process and highlights the importance of early diagnosis and treatment of this disease as it appears to have greater benefit when administered early in the course of the disease. No significant difference in clinical outcomes were seen between the 20 mg orally per day and the 80 mg orally per day dosing of tafamidis. Although the trial was designed with the requirement of tissue biopsy to make the diagnosis, the use of technetium labeled bone avid radiotracers in diagnosing TTR-CA have been validated as an accurate method for identifying these patients with high sensitivity and specificity3,8,9. This non-invasive method of diagnosing this disease leads to earlier identification and therefore earlier treatment of these patients with TTR-CA. Tafamidis is the first drug approved by the Food and Drug Administration to treat patients with TTR-CA related cardiomyopathy, this drug was approved May 3, 2019.
Diflunisal is a non-steroidal anti-inflammatory drug that binds and stabilizes the transthyretin protein in TTR-CA against acid mediated fibril formation. Dosing is 250 mg po twice daily, side effects include cyclooxygenase (COX) enzyme related fluid overload, nephrotoxicity and gastrointestinal bleeding. This drug is still in trial phase and not yet approved by the FDA for use in patients with TTR-CA10.
(iii) TTR Fibril Degradation and Absorption
Fibril degradation and reabsorption in patients with TTR-CA can be achieved with several therapeutic agents such as doxycycline-tauroursodeoxycholic acid (TUDCA) and monoclonal anti-serum amyloid protein (SAP) antibody10.
Doxycycline-TUDCA removes amyloid protein that is already deposited and is administered orally as 100 mg BID/250 mg TID and is still under investigation10.
Monoclonal anti-serum amyloid protein (SAP) antibody works as an antibody against a normal non-fibrillar glycoprotein SAP and promotes a giant cell reaction that removes visceral amyloid deposits and is administered intravenously and is still under investigation. Potential side effects are infusion site reactions10.
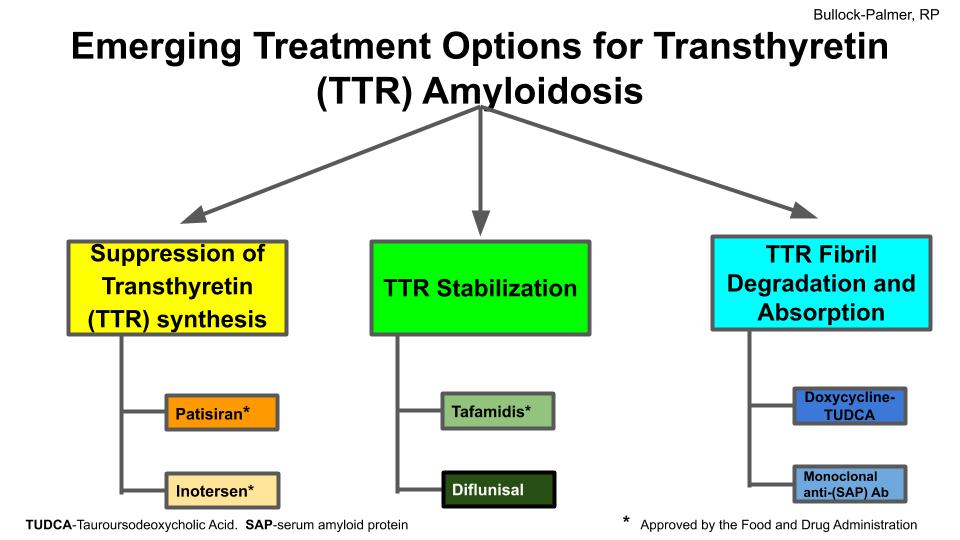
Suppression TTR in reference 5 and 6. TTR stabilization and TTR fibril degradation/absorption in reference 10.
Monitoring Treatment:
With emerging therapeutic options for patients with TTR-CA, there is a need for a reliable method for detecting disease progression and monitoring improvement of the disease with treatment. While 99mTc-PYP has been shown to be able to accurately diagnose TTR-CA it has not been shown to be a useful tool to monitor disease progression. However, quantitative echocardiography and global longitudinal strain as well as cardiac magnetic resonance imaging (CMR) with tissue characterization as well as amyloid imaging with Positron Emission Tomography (PET) radiotracers show promise as being potential tools to monitor disease progression and monitor treatment effects but are yet to be validated11.
Conclusion
The emergence of new treatment options for patients with TTR-CA provides a great degree of hope for these patients with a disease that was once thought to be very difficult to diagnose and even more difficult to treat. The FDA’s approval of patisiran, inotersen and most recently tafamadis as outlined previously is quite exciting news for patients with TTR-CA. However, it is hopeful that there will be an opportunity to have these medications be more affordable and accessible for these patients with TTR-CA so that they can all benefit from these therapies to improve their clinical outcomes.
References:
- Bullock-Palmer RP. Diagnosing cardiac amyloidosis: A wealth of new possibilities with nuclear cardiac imaging. J Nucl Cardiol. 2019 May 13. Doi: 10.1007/s12350-019-01740-w. [Epub ahead of print] PMID: 31087262
- Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, Plana JC, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012;98(19):1442-8.
- Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016;133(24):2404-12.
- Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: Emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 2015;20(2):163-78.
- Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul 5;379(1):11-21.
- Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul 5;379(1):22-31.
- Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al.Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy.. N Engl J Med. 2018 Sep 13;379(11):1007-1016.
- Castano A, Haq M, Narotsky DL, et al. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol 2016;1:880-889.
- Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013;6:195-201.
- Castano A, Narotsky D and Maurer MS, Emerging Therapies for Transthyretin Cardiac Amyloidosis Could Herald a New Era for the Treatment of HFPEF. https://www.acc.org/latest-in-cardiology/articles/2015/10/13/08/35/emerging-therapies-for-transthyretin-cardiac-amyloidosis.
- Singh V, Falk R, Di Carli MF, Kijewski M, Rapezzi C, Dorbala S. State-of-the-art radionuclide imaging in cardiac transthyretin amyloidosis. J Nucl Cardiol 2019;26(1):158-73.