In my previous blog posts, I started to discuss the importance of toll-like receptor 4 (TLR4) and how it contributes to aortic aneurysms and how microbiota-derived lipopolysaccharide (LPS) can activate the whole signaling pathway. In today’s post, I thought to discuss the TLR4 pathway, in far more details and how it contributes to obesity and metabolic disturbances.
Production of LPS and secretion from intestinal epithelial cells results in LPS binding to cytokine receptors on hepatocytes/adipocytes and as a consequence, activation of a network of signaling pathways1. Upon binding of TRL4 to its co-receptor, myeloid differentiation factor 2 (MD2), a molecular complex is formed at surface level that becomes the binding site of LPS. LPS forms a complex with lipoprotein binding protein (LBP) that binds to cell surface CD142. Upon binding of LPS and its co-receptor CD14, the subsequent transfer of LPS to the TRL4-MD2 complex starts a cascade of events leading to the activation of transcription factors that enhances the expression of many proinflammatory cytokines. TLR4-MD2 complex signals through two major pathways: myeloid differentiation factor 88 (MyD88) and TIR domain containing adaptor-inducing IFNβ (TRIF; also known as TICAM1)2. Upon ligand recognition in the MyD88 dependent pathway, it is recruited to the cytoplasmic domain of TLR. Then, protein families of TNF-α receptor associated factor 6 (TRAF6), IL-1 receptor associated kinase 1 (IRAK1) and IRAK2 are recruited by MyD883. TRAF6 activates the transforming growth factor β-activated kinase 1 (TAK1) which promotes phosphorylation of kappa beta kinase (IKK) inhibitors α, β and γ. Phosphorylated IKK complex leads to the degradation of inhibitory kappa B (IκB) and as a consequence, translocation of NFκB to the nucleus resulting in the induction of proinflammatory cytokines4. Severe reactions to the LPS are attributed to the MyD88 activation pathway resulting in production of IL-12, IL-6 and TNF-α 2. Activation of TRIF (or MyD88 independent) pathway occurs after endocytosis of TLR4-MD2 complex and is characterized by the activation of mitogen activated protein kinases (MAPKs) such as p38, ERK1/2 and c-Jun N-terminal Kinases (JNK). In the independent pathway, the induction of IFNβ and IFN inducible proteins such as monocyte chemoattractant protein 1 (MCP-1 also known as CCL2), IFNγ-induced protein (IP10 also known as CXCL10) and RANTES (also known and CCL5) are triggered5. Cani group’s study in CD14 deficient mice showed that HFD or administration of LPS showed no effect on any parameters of metabolic syndrome symptoms, further suggesting a role for TLR4 in mediating metabolic endotoxemia, adiposity and insulinemia6. Another study confirmed the aforementioned suggestion by showing a less effect on adiposity of TLR4 deficient mice challenged with HFD7. The authors also reported a higher LPS content in the cecal samples in HFD mice compared to LFD, with a close link to TLR4 induction and NFκB activation. The latter induced the expression of iNOS and COX2 while HFD challenged TLR4 deficient mice did not show activation of NFκB and changes in the mRNA levels of proinflammatory cytokines. It is also worthy to mention that the metabolic endotoxemia induced by LPS is associated with insulin resistance by activation of JNK8. This activation has the potential to promote phosphorylation of insulin receptor substrate 1 (IRS-1) at serine sites which may inhibit the normal signal transduction through insulin receptor/IRS-1 axis resulting in insulin resistance9. In addition, activation of signaling cascade induced by LPS-TLR4 increases the expression of inducible nitric oxide synthase10. The latter reacts with cysteine residues to form adducts of S-nitrosothiols which inhibits insulin signal transduction via phosphorylation of IRS-1 in serine leading to insulin resistance in hepatic, muscle and adipose tissue10.
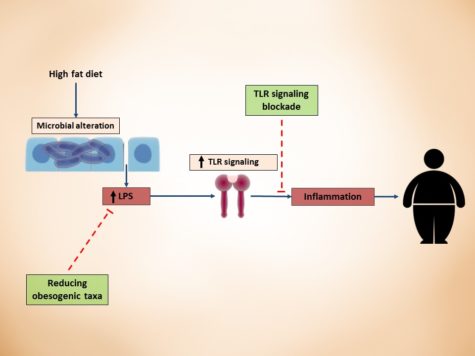
How LPS-derived metabolic endotoxemia results in obesity onset. A high fat diet can result in a shift in gut microbiota composition which can contribute to increased gut permeability and metabolic endotoxemia. The gut microbiota-derived LPS activates TLRs which produce proinflammatory cytokines that can contribute to onset of obesity. Abbreviations: LPS, lipopolysaccharide; TLR, toll like receptor.
In a recent study on both Myd88-/- and Trif-/- mice by Fredrik Backhed group, authors investigated the effect of lard diet (rich in saturated lipids) on gut microbiota composition compared with fish oil fed (enriched in polyunsaturated fatty acids) mice11. Their result demonstrated that mice lacking MyD88 and TRIF are protected against lard-induced white adipose tissue (WAT) inflammation and metabolic perturbations and the saturated dietary lipids interact with gut microbiota to induce inflammation in WAT. Authors reported that lard fed mice showed an increase in the serum levels of LPS compared to fish oil fed group, indicating that microbial factors may be present in the periphery that may affect WAT inflammation. Moreover, they showed that fish oil diet had increased the levels of taxa from the genera Lactobacillus and Akkermansia. Also, studies on Akkermansia muciniphila have been shown a reduction in fat mass gain and WAT macrophage infiltration alongside improvement of gut barrier function when administered to mice with HFD-induced obesity12. Microbiota transplantation from fish oil fed mice into antibiotic treated mice also showed an increase in the levels of Akkermansia with partial protection against adiposity and inflammation after 3 weeks of lard diet11.
Additionally, Vijay-Kumar and his colleagues have shown that TLR5-deficinet C57Bl/6J mice exhibit hyperphagia and develop characteristics of metabolic syndrome such as increased adiposity, insulin resistance and hyperlipidemia13. They reported that loss of TLR5 and the observed metabolic changes correlated with microbiota compositional changes and induction of inflammatory signaling. TLR5 is a component of innate immune system that is expressed in the gut mucosa and flagellated bacteria can interact with TLR5 to induce activation of pro-inflammatory gene programs for host protection14.
Taken all together, current data suggest that intestinal inflammation could be the early consequence of HFD and may induce obesity via increased levels of LPS, suggesting a causative role for gut inflammation in the onset of obesity. In addition, TLR4 is the primary receptor mediating the proinflammatory effects of LPS, therefore regulating levels of LPS and/or ligand binding capacity of TLR4 may be a target to stop progression of obesity and metabolic syndrome.

Shayan is a caffeine-dependent Ph.D. Candidate at the Saha Cardiovascular Research Center, University of Kentucky. His research area is focused on vascular biology and lipid metabolism. He tweets @MoradiShayan, blogs at shayanmoradi.com and he is the Winner of World’s Best Husband Award (Category: nagging).
References:
- Park DY, Ahn YT, Park SH, Huh CS, Yoo SR, Yu R, Sung MK, McGregor RA, Choi MS. Supplementation of lactobacillus curvatus hy7601 and lactobacillus plantarum ky1032 in diet-induced obese mice is associated with gut microbial changes and reduction in obesity. PLoS One. 2013;8:e59470
- Needham BD, Trent MS. Fortifying the barrier: The impact of lipid a remodelling on bacterial pathogenesis. Nat Rev Micro. 2013;11:467-481
- Akashi-Takamura S, Miyake K. Tlr accessory molecules. Current Opinion in Immunology. 2008;20:420-425
- Kawai T, Akira S. Tlr signaling. Cell Death Differ. 2006;13:816-825
- Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: Focus on the toll-like receptors. Journal of Internal Medicine. 2007;261:511-528
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti J-F, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761-177
- Kim K-A, Gu W, Lee I-A, Joh E-H, Kim D-H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the tlr4 signaling pathway. PLoS ONE. 2012;7:e47713
- Khan Muhammad T, Nieuwdorp M, Bäckhed F. Microbial modulation of insulin sensitivity. Cell Metabolism. 2014;20:753-760
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116:1793-1801
- Sugita H, Kaneki M, Tokunaga E, Sugita M, Koike C, Yasuhara S, Tompkins RG, Martyn JAJ. Inducible nitric oxide synthase plays a role in lps-induced hyperglycemia and insulin resistance. American Journal of Physiology – Endocrinology and Metabolism. 2002;282:E386-E394
- Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani Patrice D, Bäckhed F. Crosstalk between gut microbiota and dietary lipids aggravates wat inflammation through tlr signaling. Cell Metabolism. 2015;22:658-668
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proceedings of the National Academy of Sciences. 2013;110:9066-9071
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science. 2010;328:228-231
- Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR. Deletion of tlr5 results in spontaneous colitis in mice. J Clin Invest. 2007;117